Purpose of implementing a device identification system is to improve patient safety by making the traceability of devices more efficient and simplify a FSCA (Field Safety Corrective Action) e.g. recall of devices. In parallel, the post-market safety-related activities for devices will also be more robust and allow a better monitoring by the competent authorities.
UDI, Unique Device Identifier, is a unique numeric or alphanumeric code that relates to a specific medical device. UDI allows a clear and unambiguous identification of a specific device on the market and facilitates traceability during it’s entire lifecycle. Currently there are 4 issuing entities that are designated to provide manufacturers with a list of UDIs to assign on their medical devices[1]
The UDI combines different information about the device itself and the legal manufacturer information. Purpose of implementing this device identification system is to improve patient safety by making the traceability of devices more efficient and simplify a FSCA (Field Safety Corrective Action) e.g. recall of devices. In parallel, the post-market safety-related activities for devices will also be more robust and allow a better monitoring by the competent authorities. UDI is an addition to, and not a substitute for, the existing labelling information and applicable requirements.
The legal manufacturer shall assign the UDI to the device and to all higher levels of packaging before placing a device on the market. The UDI carrier shall be placed on the label of the device and on all higher levels of packaging. Within the EU, the manufacturer also need to assign a Basic UDI-DI. The Basic UDI-DI is the main key for device-related information entered in European medical device database (EUDAMED). The Basic UDI-DI shall be included in relevant documentation e.g. certificates, declaration of conformity, technical documentation and summary of safety and clinical performance.
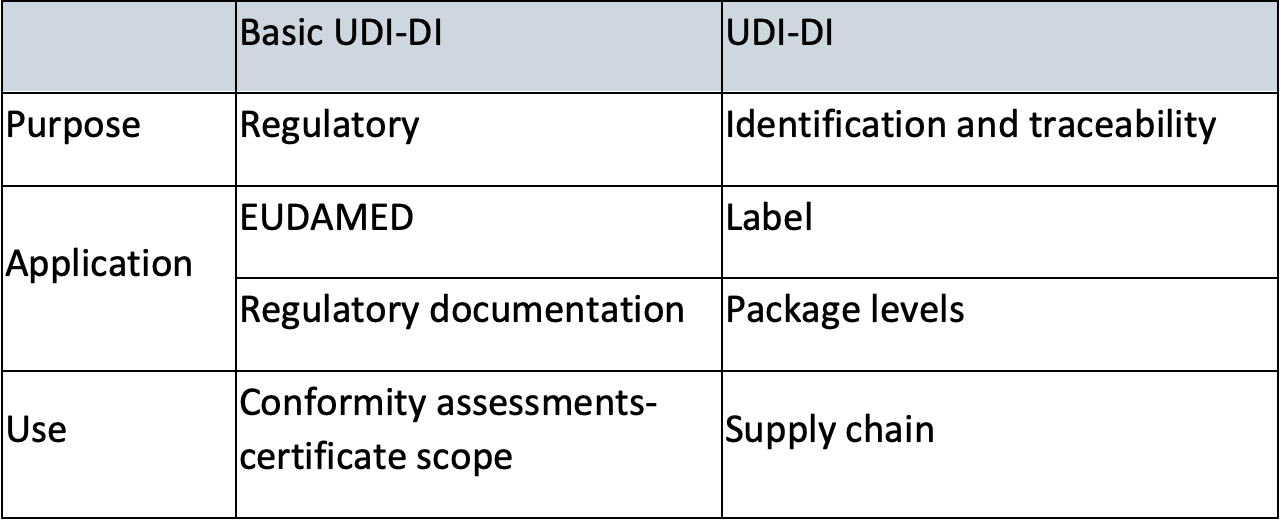
Basic UDI-DI
A Basic UDI-DI is intended to identify and connect devices with the same intended purpose, risk class and essential design and manufacturing characteristics. It is independent/separate from the packaging/labelling of the device and it does not appear on any trade item.
The Basic UDI-DI shall not be confused with the UDI-DI.
Key differences between Basic UDI-DI and UDI-Di are clarified in below table[2]
[1] https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32019D0939
[2] https://compliancenavigator.bsigroup.com/en/medicaldeviceblog/overview-of-the-eu-basic-udi-di
A new UDI shall be assigned whenever there is a change that could lead to misidentification of the device and/or ambiguity in its traceability, in particular in following changes.
- name or tradename
- device version model
- labelled as single use
- packaged sterile
- need for sterilization before use
- quantity of devices provided in the packaging
- critical warnings, contraindication
UDI-DI and UDI-PI
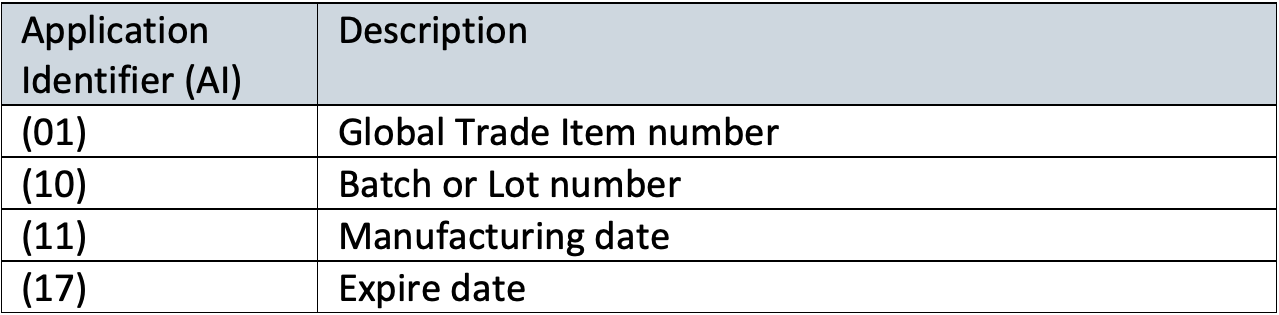
The difference between UDI-DI (device identifier) and UDI-PI (production identifier) is the type of information that need to be included.
- UDI-DI is a fixed code that relates to a specific version or model of a device.
- UDI-PI, is a variable code that relates to production data of the device, such as lot/batch number, expiry date, manufacturing date, etc.
The UDI-carrier, Automatic Identification and Data Capture (AIDC) and Human Readable Interpretation (HRI) shall be placed on the label or on the device itself and on all higher level of packaging.
A UDI consists of a combination of a Device Identifier as unique identifier (product code/GTINs) and one or more Production Identifiers (Application Identifiers (AIs)).
Example:
The UDI must appear in a plain-text version/human readable information (HRI) and in a form that uses AIDC technology. AIDC means any technology that conveys the unique device identifier or the device identifier of a device in a form that can be entered into an electronic patient record or another computer system via an automated process. HRI consists of legible characters that can easily be read by people.
Labelling specification
To ensure that your medical device fulfill the labelling requirements defined in the MDR and IVDR regulations you should start with development of a procedure there you define applicable requirements for your organization and for the type of product you place or intend to place on the market. The procedure should also define which type of information (product/packaging label, Instruction for Use (IFU), maintenance/service information and marketing material) you need to supply with your device and how it need to be supplied. Remember that the supplied information, including information for safety, are related to the result from the performed risk assessment.
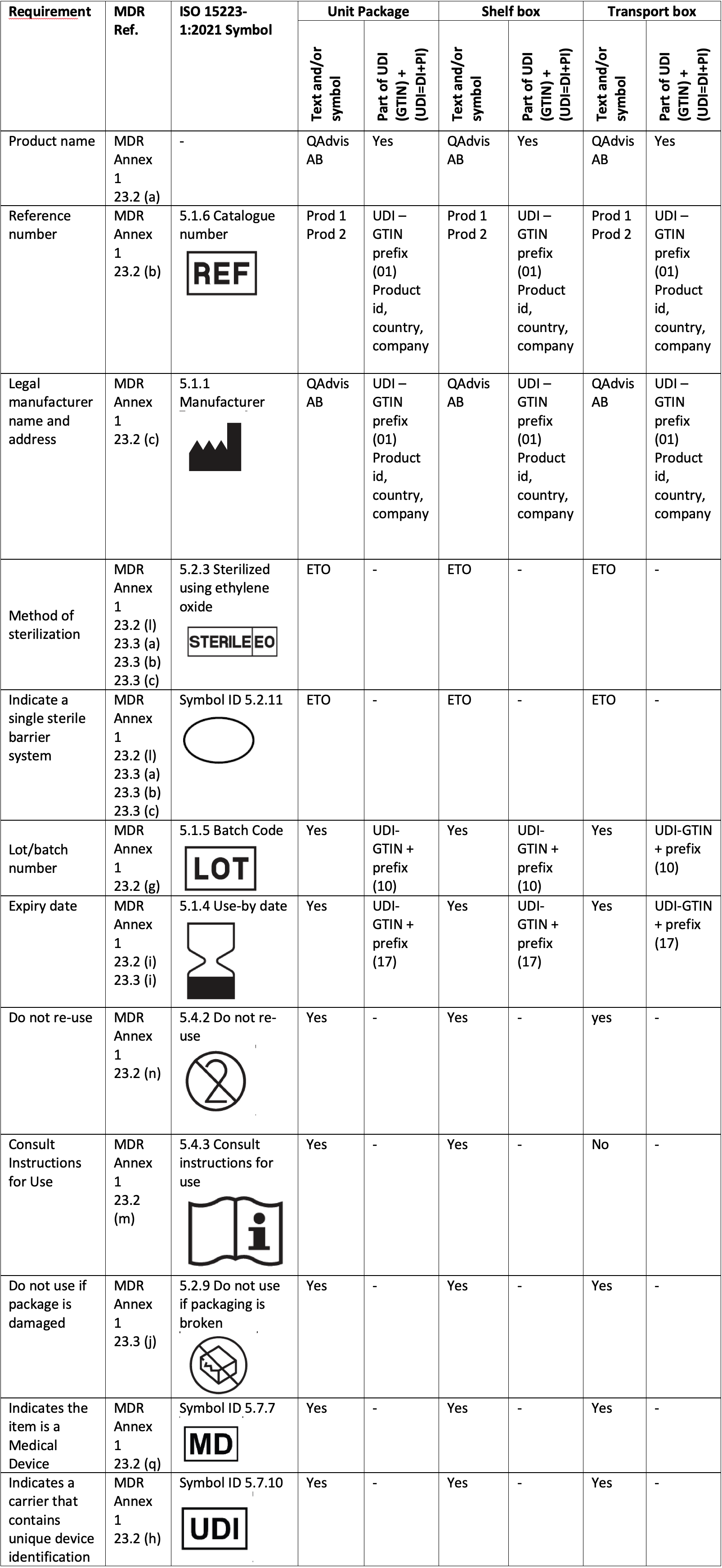
Below table is an example of how to document MDR requirements, packaging levels and how it can be related to the UDI-DI and UDI-PI.
Have in mind that countries outside Europe for example USA, Kina, Korea and Saudi Arabia also require UDI, but they may not accept all UDI issuer entities as in Europe.
If you have any question or need support with writing your procedure and/or to identify applicable labelling requirements for your organization and your device(s), just send me an email, annelie.hagstrom@qadvis.com
See https://webgate.ec.europa.eu/udi-helpdesk/?lang=enEU for more information from the EU Commission.

 We are delighted and proud to announce that QAdvis Group has achieved ISO 13485:2016 certification. This is a major milestone and demonstrates our commitment to provide high quality consulting services and we are now better positioned to consistently meet and exceed our clients’ expectations.
We are delighted and proud to announce that QAdvis Group has achieved ISO 13485:2016 certification. This is a major milestone and demonstrates our commitment to provide high quality consulting services and we are now better positioned to consistently meet and exceed our clients’ expectations.